Molecular Dynamics (MD) simulation is a key tool for the computational exploration of the dynamic and
equilibrium properties of molecular systems. However, computational studies of large systems such as biological macromolecules pose a significant challenge
because of their size and wide range of characteristic time scales (from picoseconds for chemical bond fluctuations to years for molecular aggregation).
For example, state-of-the art MD software has only recently achieved microsecond-scale simulations of relatively small
proteins (~100 residues), employing hundreds or thousands of CPUs to do so. The πDMD software package is designed to perform simulations of large systems at
long time-scales while utilizing computational minimal resources.
WHAT DOES IT DO?
πDMD is a high-performance parallelized software package that implements an efficient event-driven algorithm (Discrete Molecular Dynamics, DMD) to reach
relevant time and length scales while occupying minimal computational resources. The πDMD software package includes the Medusa force field, an implicit
solvent force field benchmarked on proteins, polynucleotides, and small organic molecules. The Medusa force field allows for the efficient implementation of
specific interactions, such as hydrogen bonds, greatly accelerating the speed of simulation. The πDMD simulation engine also supports canonical and
micro-canonical ensemble simulations utilizing a User-specified force field and/or constraints. Using πDMD, the user can observe the evolution of the
system over time. πDMD allows for tracking the coordinates of each particle or atom, as well as the total energy and system temperature and pressure.
To improve the sampling of quenched systems, the πDMD software package includes support for the replica exchange protocol as well as single-trajectory
simulations.
ADVANTAGES
The πDMD simulation engine is extremely fast and efficient when compared to its peers; using πDMD, one can achieve the folding of a small protein
on a single desktop machine in less than a week. With a small cluster of 30 CPUs, several protein folding and unfolding events can be observed in a similar
time frame. A similar problem recently addressed with a conventional MD engine required the use of lengthy simulations performed on an expensive,
privately-owned machine designed specifically for MD simulations. πDMD provides a reasonably priced software package capable of extensive
conformational sampling, tested and proven on challenging and biologically relevant systems and problems such as metallo-enzymes, peptide docking, and
protein engineering (Applications). πDMD features an enhancement of performance due to the use of implicit solvent. In contrast with MD simulations,
the πDMD energy functions are discretized in space instead of time, allowing an improved treatment of specific interactions. The πDMD software
package is conducive for use by both beginners and advanced users, providing benchmarked and proven defaults while allowing discretionary control over
simulation parameters and potentials.
- Fast and efficient in comparison with peers; provides extensive conformational sampling to a level matched only by expensive privately-owned
specialty machines.
- πDMD may be run on a desktop/notebook computer; no need for specialized equipment.
- Tested and experimentally proven on challenging and biologically relevant systems.
- Use of implicit solvent enhances simulation speed while improved treatment of specific interactions permits equivalent performance to
explicit solvent MD simulations of biomolecules.
- Molecular simulation tool conducive for use by both beginners and advanced users.
APPLICATIONS

Protein folding
DMD can be used to reproduce the folding pathway of proteins. Khare et al. reproduced the experimental folding behavior of Cu, Zn Superoxide Dismutase (SOD1)
(Khare et al.,
Journal of Molecular Biology, 334: 515-525 (2003)). Using the DMD simulations, Khare et al. found that the residues most important
for protein stability are also those responsible for the two-state folding of the SOD1 monomer, and that several of these residues are associated
with documented disease-causative mutations. Proctor et al. studied the thermodynamic and structural effects of post-translational modifications to SOD1
using both replica exchange and equilibrium DMD simulations (Proctor et al.,
Journal of Molecular Biology, 408: 555-567 (2011)). Using DMD simulations,
Proctor et al. identified a mechanism by which glutathionylation increases the dissociation of the SOD1 dimer: steric clashing caused by glutathione
changes the number and composition of interactions in the dimer interface, resulting in the experimentally-observed dimer dissociation.
Structural refinement
πDMD can be used to eliminate clashes and reconstruct insertions and deletions for the construction of homology models. Serohijos et al. have
utilized the DMD method in their model of the Cystic Fibrosis Transmembrane Regulator (CFTR) for the modeling of the R domain (Serohijos et al.,
Proceedings of the National Academy of Sciences USA, 105: 3256-3261 (2008)). Ramachandran and Kota et al. used DMD to resolve clashes in
protein structural models with minimal perturbation to the protein backbone (Ramachandran et al.,
Proteins, 79: 261-270 (2011)).
Benchmark studies of this structure resolution method demonstrate its efficiency and accuracy in comparison with other resolution methods.
Identifying ligand binding pose
The docking and virtual screening of small molecules is an integral part of drug design in both industry and academia. Proctor et al.
utilized DMD to identify native or near-native binding poses of small molecule ligands in difficult virtual screening targets, which could not be
distinguished with several popular scoring functions such as AutoDock and Glide (Proctor et al.,
Biophysical Journal 102: 144-151 (2012)).
The authors found that the average simulation time that the ligand spent in the pose over many simulations could be used as a statistical measure
to incorporate entropy and protein-ligand dynamics into the identification of ligand binding pose.
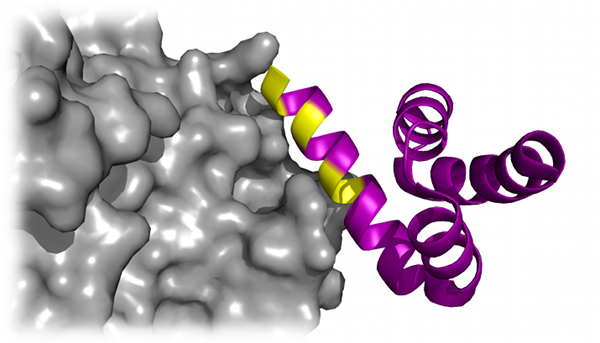
Peptide docking
Using the powerful sampling ability of DMD, Dagliyan et al. recapitulate the native binding sites and native-like binding poses of
protein-peptide complexes (Dagliyan et al.,
Structure, 19: 1837-1845 (2011)). The efficiency of the DMD algorithm allows for extensive
sampling of the protein and peptide conformational space, along with side chain stability, which the authors found was a crucial factor in
determining the native binding site of the peptide.
Protein design
Jha et al. used the conformational sampling abilities of DMD to enhance sequence design protocols for the redesign of a scaffold protein to bind
to the kinase domain of PAK1 (Jha et al.,
Journal of Molecular Biology, 400: 257-270 (2010)). The authors experimentally confirmed the
designed protein to have micro-molar binding affinity for PAK1, and that the designed protein bound the target at the intended site. This design
of novel protein-protein interactions has implications for the design of therapeutics and molecular probes.
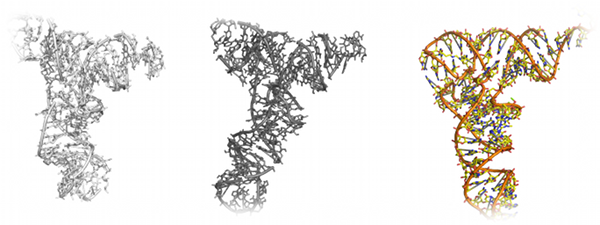
RNA folding and structure determination
The folding of RNA molecules is an extremely challenging task due to the many degrees of freedom of each RNA base. RNA is a more flexible molecule
than are proteins, creating a more extensive conformational space. For this reason, few studies have attempted ab initio folding of RNA. Ding et al.
developed a simplified three-bead model for RNA molecules, and utilized this model to successfully fold 150 structurally diverse RNA sequences from
the fully extended conformation (Ding at al.,
RNA, 14: 1164-1173 (2008)). To fold larger RNAs, such as tRNA, Gherghe et al. incorporated
experimental knowledge of the RNA secondary structure into simulation constraints in order to decrease the size of conformational space during
folding (Gherghe et al.,
Journal of the American Chemical Society, 131: 2541-2546 (2009)). The authors use a coarse-grained RNA model of DMD
along with constraints derived from SHAPE analysis to fold yeast tRNA
Asp to 3.8 Å of the crystal structure. This hybrid experimental and
computational approach demonstrates the power of DMD for use in the structure determination of biological molecules.
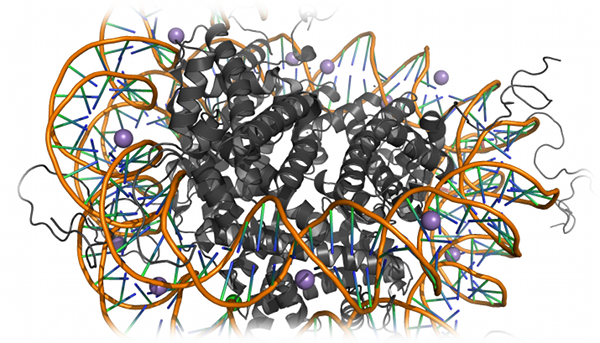
Nucleosome dynamics
Nucleosome dynamics is crucial for the regulation of chromatin stability and, hence, gene regulation. Sharma et al. utilized DMD to build a structural
model of the nucleosome using a coarse-grained model of DNA and histone proteins (Sharma et al.,
Biophysical Journal, 92: 1457-1470(2007)).
The authors used this model to reconstruct all-atom structures highlighting DNA-histone interactions, and found that histone tails may have a crucial
role in chromatin stability, via salt-bridge interactions with DNA.
Multi-scale modeling
In large systems with many degrees of freedom, multi-scale modeling can be used to provide high resolution to areas of interest, and lower
resolution elsewhere. Ding et al. use a multi-scale approach to model the aggregation of mutant and wild type SOD1 (Ding et al., Journal of
Molecular Biology, in press, (2012)). The authors found that mutations to SOD1 alter the unfolding pathway of SOD1 monomers, causing
differences in morphology in the resulting aggregates. Their results implicate local unfolding in SOD1 aggregation, suggesting a new therapeutic
target for ALS.
Liquids and artificial materials
πDMD is useful not only in biological systems, but in any system with many degrees of freedom. Buldyrev et al. utilized the DMD approach to study
the dynamics of water-like solutes (Buldyrev et al., Proceedings of the National Academy of Sciences USA, 104: 20177-20182 (2007)). Water,
although a seemingly simple molecule, is a challenge to model properly, and a large field has grown around the attempt to simplify its representation
in simulations. The authors describe a simplified water model that successfully exhibits water-like hydration properties, and use DMD to parameterize
this model. The efficient calculation and sampling of the DMD method can also be employed for engineering applications. For example, Schultz et al.
utilized DMD to study the phase behaviors of nanoparticles in solution with copolymer (Schultz et al., Macromolecules, 38: 3007-3016 (2005)).
The authors’ results revealed important behaviors in polymer localization and spacing, critical to the function of these nanomaterials.
REFERENCES
N. V. Dokholyan, S. V. Buldyrev, H. E. Stanley, and E. I. Shakhnovich, “Molecular dynamics studies of folding of a protein-like model.” Folding and
Design, 3:577-587 (1998)
F. Ding, D. Tsao, H. Nie, and N. V. Dokholyan, “Ab initio folding of proteins with all-atom discrete molecular dynamics.” Structure,
16:1010-1018 (2008)
E. A. Proctor, F. Ding, and N. V. Dokholyan, “Discrete Molecular Dynamics.” Wiley Interdisciplinary Reviews: Computational Molecular Science,
1:80-92 (2011)
D. Shirvanyants, F. Ding, D. Tsao, S. Ramachandran, and N. V. Dokholyan, “DMD: an efficient and versatile simulation method for fine protein
characterization.” Journal of Physical Chemistry, in press (2012)
SUPPORT
For support contact Molecules in Action via email or phone.