
Comparative modeling of protein structure based on a homolog of known structure offers the most viable alternative to experimental structure determination. However, achieving the quality of the homology model comparable to experimental structures is a challenge. MIA suite provides two programs, Chiron and Loop, that harness the efficient sampling capability of the physically accurate all-atom DMD simulations to ensure your homology models are of structural quality comparable to experimental structures.
Loop. In the pipeline of homology model building, adding loops to the protein structure is an important and technically challenging step. The loop module utilizes programs that automate DMD for the specific purpose of loop-building.
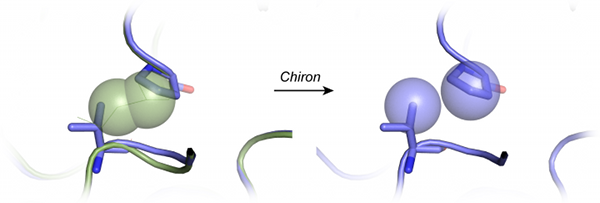
Chiron. Chiron is the first program in its class that utilizes an energetic measure for steric clashes: the "clash-score". Moreover, by using a distribution of the "clash-score" of high-resolution structures, Chiron can report the Z-score of a given structure in terms of its clash-score. Once it has been identified that a given structure features unacceptable Z-score, Chiron uses DMD to minimize the clash-score. DMD is optimized such that, using specific parameters, Chiron can be used to resolve even severe clashes that cause traditional MD to fail. Additionally, Chiron causes minimal perturbation (less than 1 Å Cα RMSD in a benchmark set of structures with severe clash-scores). Thus, it is the ideal tool to ensure homology models have physiologically acceptable clash-scores.
- Chiron can minimize structures with severe clashes, where other methods fail.
- In benchmark tests, Chiron is able to resolve clashes from the homology models within 1Å of the initial model and yet not drift away from the native structure.
- Chiron is the first program to use an energetic measure for steric clashes, which is a more realistic way to evaluate quality of a protein structure
 Robust Clash Minimization
Robust Clash Minimization
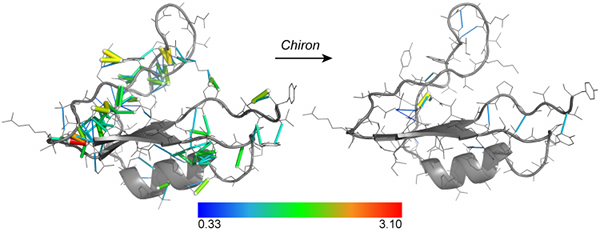 Chiron works where traditional MD fails
Chiron works where traditional MD fails
Chiron: S. Ramachandran, P. Kota, F. Ding, and N. V. Dokholyan, "Automated minimization of steric clashes in protein structures." PROTEINS: Structure, Function and Bioinformatics, 79: 261-270 (2011)
Gaia: P. Kota, F. Ding, S. Ramachandran, and N. V. Dokholyan, "Gaia: Automated quality assessment of protein structure models." Bioinformatics, 27: 2209-2215 (2011)