Eris, which takes the name of Greek goddess of discord, is protein stability prediction and design
software. Using Eris, one can rapidly estimate a protein’s stability changes upon mutations, which are often introduced to manipulate its original function
or to create new functions. A priori knowledge of stability changes can help both to avoid laborious exploratory experiments and to design stable
and functional proteins. Using Eris, one can also design protein sequences that increases a protein’s stability, which is required in the development
of protein therapeutics, such as antibodies.
WHAT DOES IT DO?
Using Eris, one can calculate the change of the protein stability induced by mutations (Delta−Delta−G, ΔΔG) utilizing the Medusa modeling suite.
Given a protein structure and single or multiple mutations as input to Eris, the user can rapidly evaluate the ΔΔG values induced by such mutations.
A negative ΔΔG value suggests that the mutant is more stable than the wild type, while a positive ΔΔG value suggests that the mutations
destabilize the protein as compared to the wild type.
Using Eris, one can design a protein’s sequence to make the protein more stable. The user can choose to redesign the entire protein or a sub-set of protein
sequence positions. The latter is often necessary in order to avoid affecting the functional sites of the protein, such as enzyme active sites or binding
interfaces. The output of Eris is the optimal sequences and their associated energies.
ADVANTAGES
Eris has been benchmarked on a large set of experimentally measured ∆∆G values (>500). The mutation-induced stability changes, ΔΔG values,
were calculated and compared with the experimental data, and a significant correlation was found. The overall correlation coefficient was 0.75. In contrast
to many other stability prediction methods, Eris is not trained using experimental protein stability data, and is therefore unbiased and valid for a wide
range of proteins. Moreveor, Eris models backbone flexibility, which is crucial for ∆∆G estimation of small−to−large mutations.
- Benchmarking over a large set of experimentally measured ΔΔG values (>500 mutations), we observe observe a significant correlation between
calculated and experimental ΔΔG (0.75), demonstrating that Eris is one of the best programs of its class.
- Eris predictions are valid over a wide range of proteins because, unlike other programs, Eris has not been trained using specific
experimental protein stability data.
- The Eris protocol includes backbone flexibility, which is crucial for accurate ΔΔG estimation for mutations from a small amino acid
to a large one.
APPLICATIONS

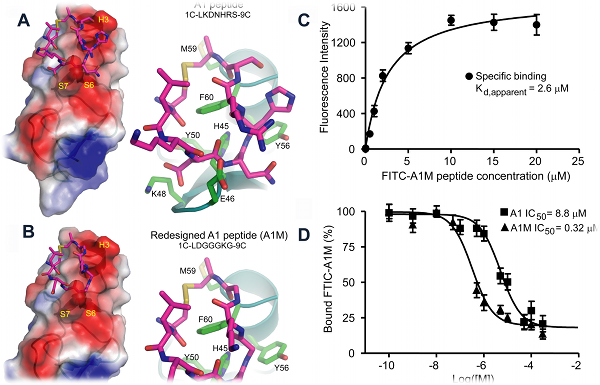
Designing a cyclic peptide that binds to CRIP1, a biomarker for breast cancer
Cysteine-rich intestinal protein 1 (CRIP1) has been identified as a novel marker for early detection of various cancers. In collaboration with
Dr. James P. Basilion’s group (Case Western Reserve University), we combined phage display with molecular modeling to identify a high-affinity
ligand for CRIP1. Panning experiments using a circularized C7C phage library yielded several consensus sequences with modest binding affinity to
purified CRIP1. Two sequence motifs having the highest affinity for CRIP1, A1 and B1, were chosen for further study. Using Eris, we identified a
novel peptide, A1M, whose affinity was predicted to be much improved. Synthesis of the peptide and saturation binding studies demonstrated
approximately a 10-20 fold improvement in the affinity of A1M compared to either A1 or B1 peptides.
Designing yeast G-protein mutants with various stabilities
The yeast heterotrimeric G-protein α subunit Gpa1 is a key negative regulator of signal transduction in response to stimulus with mating factor,
and is therefore tightly regulated. Gpa1 is one of the very few proteins whose steady-state levels are regulated by both mono- and polyubiquitination.
Whereas monoubiquitinated Gpa1 is translocated and degraded by vacuolar proteases, polyubiquitinated Gpa1 accumulates in the cytoplasm and is degraded
by the 26S-proteosome. The determinants of the two different paths of Gpa1 turnover are unknown. We hypothesized that mono- and
polyubiquitination of Gpa1 are non-redundant degradation signals that target distinct cellular pools of Gpa1, where polyubiquitination signals the
degradation of protein that is thermodynamically unstable, and monoubiquitination signals the degradation of properly folded protein as part of a
regulated protein-clearing mechanism. To test this hypothesis, we performed molecular modeling to computationally identify a series of point mutations
in Gpa1 with a gradient of putative thermo-stabilities relative to the wild type protein. We used Eris to evaluate thermo-stabilities of
point mutations. In collaboration with Dr. Henrik Dohlman (UNC-CH), we compared the levels of mono- versus polyubiquitination for each of the Gpa1 mutant
isoforms in vivo. Those point mutations predicted to have the greatest negative affect on Gpa1 stability exhibited the greatest levels of polyubiquitination.
In contrast, the observed levels of Gpa1 monoubiquitination remained constant for all isoforms. These data demonstrate that poly- and mono-ubiquitination
are non-redundant protein clearing mechanisms involved in the turnover of distinct separate cellular populations of Gpa1.
Uncovering the importance of histone stability in yeast survival
In eukaryotes, histone proteins package genomic DNA into higher order chromatin structures, which control the accessibility of
genomic DNA to various cellular machineries. The fundamental unit of eukaryotic chromatin is the nucleosome, composed of ~147 base pairs of
DNA wrapped around the histone octamer, which comprises two copies of each of the four histone proteins: H2A, H2B, H3, and H4. The histone
proteins are highly conserved in evolution, and since evolutionary pressure is associated with survival fitness, we asked if engineered mutations
resulting in thermodynamic destabilization of the histone octamer would lead to phenotypic consequences in the budding yeast
Saccharomyces
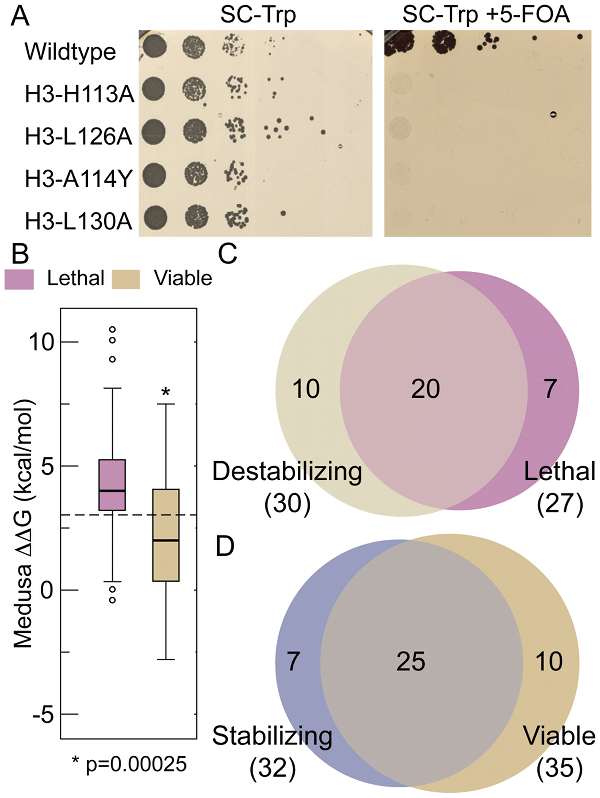
cerevisiae. Based on Eris calculations, we made four histone H3 mutations that disrupted conserved interactions (H113A, L126A, L130A and A114Y)
and destabilized the nucleosome. Consistent with these mutations resulting in a significant destabilization of the H3-H3’ interactions
in
silico, we found that these H3 mutations were lethal when engineered in budding yeast (Figure A). To extend this comparison between residues
needed for nucleosome stability and their effects on growth in yeast, we used Eris to calculate the change in nucleosome stability of 62 mutations pertaining
to buried/interface residues. We observed that residues found to be essential for viability are often also required
for nucleosome stability. The ΔΔG of lethal mutants is significantly higher than the ∆∆G of viable mutants: the probability that the
ΔΔGs of lethal and viable mutants are similar is 2.5 × 10
-4, indicating that the difference in destabilizition of lethal and
viable mutants is statistically significant. These results imply that the lethality observed in these studies is due to thermodynamic destabilization
of the nucleosome. Furthermore, if we use an arbitrary ΔΔG cut-off of +3 kcal/mol as a threshold to suggest a point where sufficient
thermodynamic destabilization would lead to lethality, we are able to predict 74% of the lethal mutants and 71% of the viable mutants from the
HistoneHits database (Figure C, D). These data suggest that nucleosome stability, which is essential for viability, is a major driving force behind
the sequence conservation of buried H3 and H4 residues.
REFERENCES
S. Yin, F. Ding, and N. V. Dokholyan, "Eris: an automated estimator of protein stability." Nature Methods 4, 466−467 (2007)
S. Yin, F. Ding, and N. V. Dokholyan, "Modeling backbone flexibility improves protein stability estimation." Structure, 15: 1567−1576 (2007)
SUPPORT
For support contact Molecules in Action via email or phone.